GeneticData
09_Tree_of_life.RmdA quick wrap-up of the git stuff from last time
We’ll install a package called usethis, which contains some helpful programming tools for the weeks ahead:
install.packages("usethis")We’ll use this to create a personal access token for github. This ensures we don’t have to retype our password every time we need to access GitHub.
usethis::create_github_token() And we will use this token to access our account via RStudio.
credentials::set_github_pat("PAT here")Now, refresh your R session.
Once this is done, we can check that this worked like so:
usethis::git_sitrep()The Tree of Life
First, we need to do some installs.
install.packages("devtools")1
devtools::install_github("ropensci/rotl")
devtools::install_github("ropensci/traits")
devtools::install_github(repo='ropensci/phylotaR', build_vignettes=TRUE)Now, load your packages:
Open Tree
First, we’re going to look for some data. Discuss with your neighbor a clade you like and want to build a tree of. For example, perhaps I really like hominids.
apes <- c("Pongo", "Pan", "Gorilla", "Hoolock", "Homo")
(resolved_names <- rotl::tnrs_match_names(apes))## search_string unique_name approximate_match ott_id is_synonym flags
## 1 pongo Pongo FALSE 417949 FALSE
## 2 pan Pan FALSE 417957 FALSE sibling_higher
## 3 gorilla Gorilla FALSE 417969 FALSE sibling_higher
## 4 hoolock Hoolock FALSE 712902 FALSE
## 5 homo Homo FALSE 770309 FALSE sibling_higher
## number_matches
## 1 2
## 2 2
## 3 1
## 4 1
## 5 1Put the names of five genera into the function. If you want, you can use mine below, or pick new ones you prefer.
ants <- c("Martialis", "Atta", "Ectatomma", "Tatuidris", "Aneuretus", "Xymmer", "Aenictus")
(resolved_names <- rotl::tnrs_match_names(ants))## search_string unique_name approximate_match ott_id is_synonym flags
## 1 martialis Martialis FALSE 334634 FALSE
## 2 atta Atta FALSE 709964 FALSE
## 3 ectatomma Ectatomma FALSE 162845 FALSE
## 4 tatuidris Tatuidris FALSE 150177 FALSE
## 5 aneuretus Aneuretus FALSE 425343 FALSE
## 6 xymmer Xymmer FALSE 4583488 FALSE
## 7 aenictus Aenictus FALSE 169919 FALSE
## number_matches
## 1 1
## 2 1
## 3 1
## 4 1
## 5 1
## 6 1
## 7 1Try to view them on a tree:
tr <- tol_induced_subtree(ott_ids = ott_id(resolved_names))##
Progress [----------------------------------] 0/43 ( 0) ?s
Progress [================================] 43/43 (100) 0s
## Warning in collapse_singles(tr, show_progress): Dropping singleton nodes
## with labels: mrcaott1682ott35311, mrcaott1682ott34821, mrcaott34821ott950983,
## Aneuretinae ott393261, mrcaott4706ott5525, mrcaott5525ott8038,
## mrcaott8038ott8685, mrcaott8685ott36581, mrcaott8685ott56535,
## mrcaott8685ott72729, mrcaott8685ott86888, mrcaott86888ott129302,
## Attini ott344333, mrcaott114767ott114779, mrcaott114779ott339738,
## mrcaott114779ott116619, mrcaott116619ott148996, mrcaott148996ott1071522,
## mrcaott148996ott150184, mrcaott148996ott339763, mrcaott148996ott409299,
## mrcaott48019ott688185, Ectatomminae ott327970, mrcaott48019ott162057,
## mrcaott7438ott5553505, mrcaott7438ott7439, mrcaott7439ott165217,
## mrcaott7439ott15430, mrcaott15430ott120483, mrcaott15430ott150187,
## mrcaott15430ott169913, mrcaott17752ott98880, mrcaott17752ott150176,
## mrcaott150176ott1037289, mrcaott150176ott4539262, Agroecomyrmecinae ott504610,
## mrcaott98886ott152924, mrcaott152924ott452719, mrcaott152924ott739132,
## mrcaott152924ott503313, mrcaott503313ott928530, mrcaott928530ott6280946,
## Martialinae ott334635
plot(tr)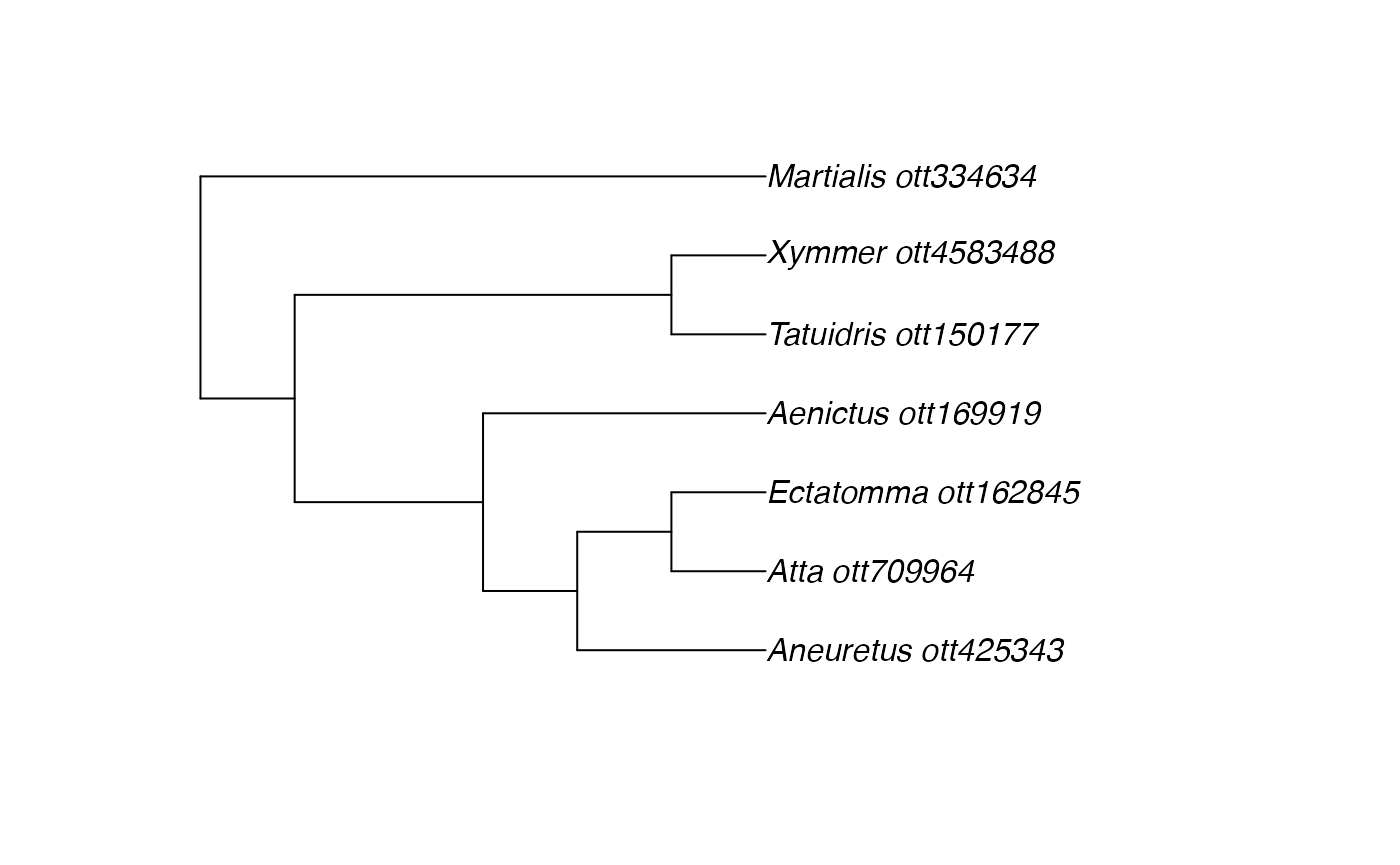
I’m a little surprised by this tree.
Looking for data
We’re going to try to build a tree of these taxa from GenBank Data. Much like accessing NOAA data, we will need to make an API Key. Go here and register.
Now that we have that key, we can access NCBI:
library(rentrez)
entrez_search(db = "Nucleotide", term="Ectatomma", api_key ="96b569c049fe3d3055c5b747112dfec84308")## Entrez search result with 679 hits (object contains 20 IDs and no web_history object)
## Search term (as translated): "Ectatomma"[Organism] OR Ectatomma[All Fields]Holy moley that’s a lot of data. If we do that for every taxon, we’ll end up with a mess. Why don’t we try to filter it a bit.
Ectatomma <- entrez_search(db = "Nucleotide", term="Ectatomma AND 2018:2019[PDAT]", retmax = 100, api_key ="96b569c049fe3d3055c5b747112dfec84308")So there is a good amount of data for our Ectatomma ants. We’re going to download some. And this gets a little tricky. First, I want you to run each line in the loop and get a feel for what each does. What does entrez_fetch do? Waht does cat do? Feel free to look at documentation.
for( seq_start in seq(1,50,100)){
recs <- entrez_fetch(db="nuccore", id = Ectatomma$ids,
rettype="fasta", retmax=50)
cat(recs, file="Ect.seqs.fasta", append=TRUE)
cat(seq_start+49, "sequences downloaded\r")
}## 50 sequences downloadedNow, let’s look at this file. What do we notice about it? What information is in this file?
Before we move on, we’re going to try one other thing. Try modifying one of the two chunks above (HINT: you will have to rerun both) in order to only get data from the mitochondrial COI gene.
All right, let’s move on to trying to write a function that will return all IDs for all of the ants.
search_year <- function(ant){
query <- paste0(ant, " AND ", "2016:2019[PDAT]")
search_returns <- c(entrez_search(db="Nucleotide", term=query, retmax=20,api_key ="96b569c049fe3d3055c5b747112dfec84308"))
return(search_returns)
}
ants <- as.vector(resolved_names$unique_name)
labels <- sapply(ants,search_year, USE.NAMES = T)Have a look at ?sapply. Talk to a partner and see if you can figure out what sapply does.
So what is the problem with this output? It’s hard to parse - let’s make it easier to pull data for one taxon at a time.
search_year <- function(ant){
search_returns = c()
query <- paste0(ant, " AND ", "2016:2019[PDAT]")
search_returns[ant] <- c(entrez_search(db="Nucleotide", term=query, retmax=20,api_key ="96b569c049fe3d3055c5b747112dfec84308"))
return(search_returns)
}
ants <- as.vector(resolved_names$unique_name)
labels <- sapply(ants,search_year, USE.NAMES = T)There’s one remaining problem, though. Have a look at your outputs. How do you think we can fix this one? Let’s have a look at when papers related to Xymmer were published:
xymmer_papers <- entrez_fetch(db="pmc", term="Xymmer", id = xymmer_papers$ids, retmax=20, rettype= "text", api_key ="96b569c049fe3d3055c5b747112dfec84308")Does this give you a hint about something that needs to change to get more data? Make the change in the below code:
search_year <- function(ant){
search_returns = c()
query <- paste0(ant, " AND ", "HINT HINT HINT:2019[PDAT]")
search_returns[ant] <- c(entrez_search(db="Nucleotide", term=query, retmax=1,api_key ="96b569c049fe3d3055c5b747112dfec84308"))
return(search_returns)
}
ants <- as.vector(resolved_names$unique_name)
labels <- sapply(ants,search_year, USE.NAMES = F)Now, we’re going to get the genetic data.
search_seq <- function(labels){
recs[ant] <- entrez_fetch(db="nuccore", id = labels,
rettype="fasta", retmax=1)
cat(recs, file="Ants.seqs.fasta", append=TRUE)
return(recs)
}
seq_returns <- sapply(labels, search_seq, USE.NAMES = T)What might be a problem with these data? Look carefully at the headers. Now, decide how to fix this. I suggest thinking about a common gene for phylogenetics, COI.
Just for fun
Take a quick look at this output - it’s a little neat. We’re not going to go into building phylogenetic trees - that’s a topic for Systematics. Here’s a quick look at these data.
install.packages("ape")## Installing package into '/private/var/folders/54/9kd8nf1x4fnft0ymvb11qmc80000gn/T/RtmppNL377/temp_libpath4e15404a9177'
## (as 'lib' is unspecified)##
## The downloaded binary packages are in
## /var/folders/54/9kd8nf1x4fnft0ymvb11qmc80000gn/T//RtmpofSZb4/downloaded_packages
coi <- ape::read.dna("Ants.seqs.fasta", format="fasta")